| MitImpact id |
MI.10758 |
MI.10759 |
| Chr |
chrM |
chrM |
| Start |
3308 |
3308 |
| Ref |
T |
T |
| Alt |
C |
A |
| Gene symbol |
MT-ND1 |
MT-ND1 |
| Extended annotation |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 1 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 1 |
| Gene position |
2 |
2 |
| Gene start |
3307 |
3307 |
| Gene end |
4262 |
4262 |
| Gene strand |
+ |
+ |
| Codon substitution |
ATA/ACA |
ATA/AAA |
| AA position |
1 |
1 |
| AA ref |
M |
M |
| AA alt |
T |
K |
| Functional effect general |
start_lost |
start_lost |
| Functional effect detailed |
start_lost |
start_lost |
| OMIM id |
516000 |
516000 |
| HGVS |
NC_012920.1:g.3308T>C |
NC_012920.1:g.3308T>A |
| HGNC id |
7455 |
7455 |
| Respiratory Chain complex |
I |
I |
| Ensembl gene id |
ENSG00000198888 |
ENSG00000198888 |
| Ensembl transcript id |
ENST00000361390 |
ENST00000361390 |
| Ensembl protein id |
ENSP00000354687 |
ENSP00000354687 |
| Uniprot id |
P03886 |
P03886 |
| Uniprot name |
NU1M_HUMAN |
NU1M_HUMAN |
| Ncbi gene id |
4535 |
4535 |
| Ncbi protein id |
YP_003024026.1 |
YP_003024026.1 |
| PhyloP 100V |
4.722 |
4.722 |
| PhyloP 470Way |
0.458 |
0.458 |
| PhastCons 100V |
1 |
1 |
| PhastCons 470Way |
0.156 |
0.156 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
0.99 |
0.99 |
| SIFT |
deleterious |
deleterious |
| SIFT score |
0.0 |
0.0 |
| SIFT4G |
Damaging |
Damaging |
| SIFT4G score |
0.0 |
0.0 |
| VEST |
Neutral |
Neutral |
| VEST pvalue |
0.07 |
0.06 |
| VEST FDR |
0.35 |
0.35 |
| Mitoclass.1 |
neutral |
damaging |
| SNPDryad |
Neutral |
Neutral |
| SNPDryad score |
0.05 |
0.79 |
| MutationTaster |
. |
. |
| MutationTaster score |
. |
. |
| MutationTaster converted rankscore |
. |
. |
| MutationTaster model |
. |
. |
| MutationTaster AAE |
. |
. |
| fathmm |
. |
. |
| fathmm score |
. |
. |
| fathmm converted rankscore |
. |
. |
| AlphaMissense |
. |
. |
| AlphaMissense score |
. |
. |
| CADD |
Neutral |
Deleterious |
| CADD score |
2.362247 |
3.387294 |
| CADD phred |
18.57 |
23.0 |
| PROVEAN |
Tolerated |
Tolerated |
| PROVEAN score |
-0.26 |
-1.86 |
| MutationAssessor |
. |
. |
| MutationAssessor score |
. |
. |
| EFIN SP |
Damaging |
Damaging |
| EFIN SP score |
0.528 |
0.516 |
| EFIN HD |
Damaging |
Damaging |
| EFIN HD score |
0.196 |
0.152 |
| MLC |
Deleterious |
Deleterious |
| MLC score |
0.7 |
0.7 |
| PANTHER score |
. |
. |
| PhD-SNP score |
. |
. |
| APOGEE1 |
Pathogenic |
Pathogenic |
| APOGEE1 score |
0.75 |
0.71 |
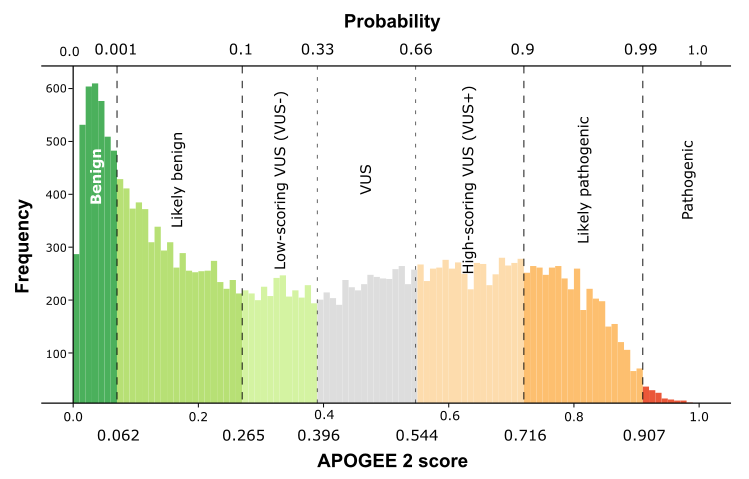
| APOGEE2 |
VUS- |
VUS |
| APOGEE2 score |
0.32785796242486 |
0.4248831060166 |
| CAROL |
deleterious |
deleterious |
| CAROL score |
1.0 |
1.0 |
| Condel |
neutral |
neutral |
| Condel score |
0.01 |
0.01 |
| COVEC WMV |
deleterious |
deleterious |
| COVEC WMV score |
4 |
4 |
| MtoolBox |
deleterious |
deleterious |
| MtoolBox DS |
0.74 |
0.76 |
| DEOGEN2 |
. |
. |
| DEOGEN2 score |
. |
. |
| DEOGEN2 converted rankscore |
. |
. |
| Meta-SNP |
. |
. |
| Meta-SNP score |
. |
. |
| PolyPhen2 transf |
. |
. |
| PolyPhen2 transf score |
. |
. |
| SIFT_transf |
. |
. |
| SIFT transf score |
. |
. |
| MutationAssessor transf |
. |
. |
| MutationAssessor transf score |
. |
. |
| CHASM |
Neutral |
Neutral |
| CHASM pvalue |
0.08 |
0.17 |
| CHASM FDR |
0.8 |
0.8 |
| ClinVar id |
9728.0 |
. |
| ClinVar Allele id |
24767.0 |
. |
| ClinVar CLNDISDB |
MedGen:CN169374|EFO:EFO_0005303,MeSH:D013398,MedGen:C0038644,OMIM:272120|MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506|MONDO:MONDO:0002032,MedGen:C0699790 |
. |
| ClinVar CLNDN |
not_specified|SUDDEN_INFANT_DEATH_SYNDROME|Leigh_syndrome|Carcinoma_of_colon |
. |
| ClinVar CLNSIG |
Benign/Likely_benign |
. |
| MITOMAP Disease Clinical info |
MELAS / DEAF enhancer / hypertension / LVNC / putative LHON |
. |
| MITOMAP Disease Status |
Reported - possibly synergistic; hg L1b and A2i marker |
. |
| MITOMAP Disease Hom/Het |
-/+ |
./. |
| MITOMAP General GenBank Freq |
0.6968% |
0.0016% |
| MITOMAP General GenBank Seqs |
426 |
1 |
| MITOMAP General Curated refs |
10720328;18335039;15466077;22777272;32094358;10521313;11938495;15972314;9299504;21968326;14960712;12160969;10070626;10924280;10371545;22777278;21041797;10519336;10803467;24002810;29987491;21625124;21457906;18194667 |
18335039 |
| MITOMAP Variant Class |
polymorphism;disease |
polymorphism |
| gnomAD 3.1 AN |
56409.0 |
56434.0 |
| gnomAD 3.1 AC Homo |
1609.0 |
0.0 |
| gnomAD 3.1 AF Hom |
0.0285238 |
0.0 |
| gnomAD 3.1 AC Het |
5.0 |
1.0 |
| gnomAD 3.1 AF Het |
8.86383e-05 |
1.77198e-05 |
| gnomAD 3.1 filter |
PASS |
PASS |
| HelixMTdb AC Hom |
1126.0 |
. |
| HelixMTdb AF Hom |
0.0057453965 |
. |
| HelixMTdb AC Het |
26.0 |
. |
| HelixMTdb AF Het |
0.00013266457 |
. |
| HelixMTdb mean ARF |
0.50148 |
. |
| HelixMTdb max ARF |
0.94643 |
. |
| ToMMo 54KJPN AC |
32 |
. |
| ToMMo 54KJPN AF |
0.000589 |
. |
| ToMMo 54KJPN AN |
54302 |
. |
| COSMIC 90 |
. |
. |
| dbSNP 156 id |
. |
. |